
Abstract
De novo mutations are known to play a prominent role in sporadic disorders with reduced fitness. We hypothesize that de novo mutations play an important role in severe male infertility and explain a portion of the genetic causes of this understudied disorder. To test this hypothesis, we utilize trio-based exome sequencing in a cohort of 185 infertile males and their unaffected parents. Following a systematic analysis, 29 of 145 rare (MAF < 0.1%) protein-altering de novo mutations are classified as possibly causative of the male infertility phenotype. We observed a significant enrichment of loss-of-function de novo mutations in loss-of-function-intolerant genes (p-value = 1.00 × 10−5) in infertile men compared to controls. Additionally, we detected a significant increase in predicted pathogenic de novo missense mutations affecting missense-intolerant genes (p-value = 5.01 × 10−4) in contrast to predicted benign de novo mutations. One gene we identify, RBM5, is an essential regulator of male germ cell pre-mRNA splicing and has been previously implicated in male infertility in mice. In a follow-up study, 6 rare pathogenic missense mutations affecting this gene are observed in a cohort of 2,506 infertile patients, whilst we find no such mutations in a cohort of 5,784 fertile men (p-value = 0.03). Our results provide evidence for the role of de novo mutations in severe male infertility and point to new candidate genes affecting fertility.
Similar content being viewed by others


Introduction
Male infertility contributes to approximately half of all cases of infertility and affects 7% of the male population. For the majority of these men the cause remains unexplained1. Despite a clear role for genetic causes in male infertility, there is a distinct lack of diagnostically relevant genes and at least 40% of all cases are classified as idiopathic1,2,3,4. Previous studies in other conditions with reproductive lethality, such as neurodevelopmental disorders, have demonstrated an important role for de novo mutations (DNMs) in their etiology5. In line with this, recurrent de novo chromosomal abnormalities play an important role in male infertility. Both azoospermia factor (AZF) deletions on the Y chromosome as well as an additional X chromosome, resulting in Klinefelter syndrome, occur de novo. Collectively, these de novo events explain up to 25% of all cases of nonobstructive azoospermia (NOA)1,4. Interestingly, in 1999 a DNM in the Y-chromosomal gene USP9Y was reported in a man with azoospermia6. Until now, however, a systematic analysis of the role of DNMs in male infertility had not been attempted, even though a pilot exome sequencing study in 13 infertile men and their parents was recently published7. This is partly explained by a lack of basic research in male reproductive health in general4,8, but also by the practical challenges of collecting parental samples for this disorder, which is typically diagnosed in adults.
In this work, we address this lack of knowledge by analysing exome sequencing data of 185 infertile males and their parents and reporting on our findings of 29 DNM in these men which are likely causative for the infertility phenotype, based on variant and gene level evidence. We emphasize an enrichment for loss-of-function (LoF) DNM in LoF-intolerant genes and missense DNM in missense-intolerant genes. We identify a number of promising candidate genes for male infertility, including the mRNA splicing gene RBM5, which contains a possibly causative DNM in our trio cohort, and possibly causative heterozygous variants in six additional patients for which parental information is not available. This work suggests a potential role for DNM as a cause of severe male infertility and addresses the need for further investigation in larger patient–parent trio cohorts to solidify these results.
Results
Discovery of de novo mutations in infertile male trios
In this study, we investigated the role of DNMs in 185 unexplained cases of oligozoospermia (<5 million sperm cells/ml; n = 74) and azoospermia (n = 111) by performing whole exome sequencing (WES) in all patients and their parents (see Supplementary Figs. 1, 2, Supplementary Notes and Data for details on methodology and clinical descriptions). In total, we identified and validated 192 rare DNMs (MAF < 0.1%), including 145 protein-altering DNMs. All de novo point mutations were autosomal, except for one on chromosome X, and all occurred in different genes (Supplementary Data 1). Two rare de novo copy-number variations (CNVs) were also identified affecting a total of 7 genes (Supplementary Fig. 3). None of the 145 protein-altering DNMs occurred in a gene already known for its involvement in autosomal dominant human male infertility. This is not unexpected as only four autosomal dominant genes have so far been linked to isolated male infertility in humans3,9.
Intolerance analysis of genes with de novo loss-of-function mutations
Broadly speaking, across genetic disorders, dominantly acting disease genes are usually intolerant to LoF mutations, as represented by a high pLI score10 or a low LOEUF score11. In our cohort of infertile men, we detected a significant enrichment in the number of LoF-intolerant genes with a LoF DNM (n = 17). No such enrichment was identified in a cohort of 1,941 control cases from de novo-db v1.6.112 (median pLI in patients with male infertility = 0.80, median pLI in controls = 3.75 × 10−5, p value = 1.00 × 10−5, N simulations = 100,000) (Fig. 1a). Similar results were obtained using the LOEUF scores (median LOEUF in patients with male infertility = 0.34, median LOEUF in controls = 0.59, p value = 1.00 × 10−5, N simulations = 100,000) (Supplementary Fig. 4a). This observation indicates that LoF DNMs likely play an important role in male infertility, similar to what is known for developmental disorders and severe intellectual disability13,14. As an example, a heterozygous likely pathogenic frameshift DNM was observed in the LoF-intolerant gene GREB1L (pLI = 1) of Proband_076. Homozygous Greb1l knockout mice appear to be embryonic lethal, however, typical male infertility phenotypic features such as abnormal fetal testis morphology and decreased fetal testis volume are observed15. Interestingly, this patient has a reduced testis volume and severe oligozoospermia (Supplementary Notes Table 1). Nonsense and missense mutations in GREB1L in humans are known to cause renal agenesis16 (OMIM: 617805), not known to be present in our patient. Of note, all previously reported damaging mutations in GREB1L causing renal agenesis are either maternally inherited or occurred de novo. This led the authors of one of these renal agenesis studies to speculate that disruption to GREB1L could cause infertility in males15. A recent WES study involving a cohort of 285 infertile men also noted several patients presenting with pathogenic mutations in genes with an associated systemic disease where male fertility is not always assessed17.

a Violin plot with quantile lines showing pLI scores in all genes in gnomAD (red), all genes affected by rare protein-altering loss-of-function (LoF) de novo mutations (DNMs) in a control population (http://de novo-db.gs.washington.edu/de novo-db/) (green) and in all genes with a rare protein-altering LoF DNM in our trio cohort (blue). Using the permutation-based, nonparametric test defined by Lelieveld et al. 64 a significant enrichment of LoF DNMs in LoF-intolerant genes in patient cohort was detected in comparison to the number of LoF in fertile control cohort (DNM LoF mutations in patients n = 17, median pLI in patients with male infertility = 0.80, DNM LoF mutations in controls n = 21, median pLI in controls = 3.75 × 10−5, p value = 1.00 × 10−5, N simulations = 100,000). The black dot indicates median pLI scores. b Violin plot with quantile lines showing the distribution of Z-scores for genes with predicted benign (n = 59) and pathogenic missense DNMs (n = 63) in infertile patients. A significant increase in predicated pathogenic DNMs in missense-intolerant genes was detected compared to benign missense DNM (Two-sided Mann–Whitney U test, p value of 3.44 × 10−4). (***p value < 0.001).
We also assessed the damaging effects of the two rare de novo CNVs by looking at the pLI score of the genes involved. Proband_066 presented with a large 656 kb de novo deletion on chromosome 11, spanning 6 genes in total. This deletion partially overlapped with a deletion reported in 2014 in a patient with cryptorchidism and NOA18. Two genes affected in both patients, QSER1 and CSTF3, are LoF-intolerant with pLI scores of 1 and 0.98, respectively. In particular, CSTF3 is highly expressed within the testis and is known to be involved in pre-mRNA 3′-end cleavage and poylyadenylation19.
Missense intolerance in de novo mutation genes
To systematically evaluate and predict the likelihood of these DNMs causing male infertility and identify novel candidate disease genes, we assessed the predicted pathogenicity of all DNMs using three prediction methods based on SIFT20, MutationTaster21, and PolyPhen222 with a minimum of 2 of the 3 showing pathogenicity to define a variant as Pathogenic. Using this approach, 84 of 145 rare protein-altering DNM were predicted to be pathogenic, while the remaining 61 were predicted to be benign. To further analyse the impact of the variants on the genes affected, we looked at the missense Z-score of all 122 genes affected by a missense variant, which indicates the tolerance of genes to missense mutations23. We identified no significant enrichment in missense DNMs in missense-intolerant genes in our infertile cohort when compared to controls (median Z-score in male infertility patients = 0.83, median Z-score in controls = 1.04, p value = 1, N simulations = 100,000) (Supplementary Fig. 4b). Interestingly, however, we observed a significantly higher median missense Z-score in genes affected by a missense DNM predicted as pathogenic (median Z-score = 1.21, n = 63) when compared to genes affected by predicted benign (median Z-score = 0.98, n = 59) missense DNMs in our cohort (p value = 5.01 × 10−4, Fig. 1b). It should be noted that the same analysis in controls showed no such significant difference (Supplementary Fig. 4c).
Protein–protein interactions reveal link to mRNA splicing
An analysis using the STRING database24, revealed a significant enrichment of protein interactions amongst the 84 genes affected by a protein-altering DNM predicted to be pathogenic (PPI enrichment p value = 2.35 × 10−2, Fig. 2). No such enrichment was observed for the genes highlighted as likely benign (n = 61, PPI enrichment p value = 0.206) or those affected by synonymous DNMs (n = 35, PPI enrichment p value = 0.992, Supplementary Fig. 5). This suggests that the proteins affected by predicted pathogenic DNMs share common biological functions.

Significantly larger number of interactions were observed in proteins affected by de novo mutations than expected for a similar sized dataset of randomly selected proteins (PPI enrichment p value = 2.35 × 10−2). The central module of the main interaction network (blue dashed circle) contains 5 proteins involved in mRNA splicing (Supplementary Fig. 6).
The STRING network analysis also highlighted a central module of interconnected proteins with a significant enrichment of genes required for mRNA splicing (Supplementary Fig. 6). The genes U2AF2, HNRNPL, CDC5L, CWC27, and RBM5 all contain predicted pathogenic DNMs and likely interact at a protein level during the mRNA splicing process. Pre-mRNA splicing allows gene functions to be expanded by creating alternative splice variants of gene products and is highly elaborated within the testis25. One of these genes, RBM5 has been previously highlighted as an essential regulator of haploid male germ-cell pre-mRNA splicing and male fertility in mice26. Mice with a homozygous ENU-induced allele point mutation in RBM5 present with azoospermia and germ cell development arrest at round spermatids. Whilst in mice, a homozygous mutation in RBM5 is required to cause azoospermia, this may not be the case in humans as is well-documented for other genes27, including the recently reported male infertility gene SYCP29. Of note, RBM5 is a tumor suppressor in the lung28, with reduced expression affecting RNA splicing in patients with non-small cell lung cancer29. HNRNPL is another splicing factor affected by a possible pathogenic DNM in our study. One study implicated a role for HNRNPL in patients with Sertoli cell-only phenotype30. The remaining three mRNA splicing genes have not yet been implicated in human male infertility. However, mRNA for all three is expressed at medium to high levels in human germ cells and all are widely expressed during spermatogenesis31. Specifically, CDC5L is a component of the PRP19-CDC5L complex that forms an integral part of the spliceosome and is required for activating pre-mRNA splicing32, as is CWC2733. U2AF2 plays a role in pre-mRNA splicing and 3′-end processing34. Interestingly, CSTF3, one of the genes affected by a de novo CNV in Proband_066, affects the same mRNA pathway18.
DNMs uncovering recessive disease and analysis of maternally inherited mutations
Whilst DNMs most often cause dominant disease, they can contribute to recessive disease, usually in combination with an inherited variant on the trans allele. In order to look for this, we analysed all DNM genes for the presence of inherited mutations on the other allele in the same patient. In Proband_060, who carried a DNM in Testis and Ovary Specific PAZ Domain Containing 1 (TOPAZ1) on the paternal allele, we did identify a maternally inherited variant predicted to be pathogenic (Supplementary Fig. 7). TOPAZ1 is a germ cell-specific gene which is highly conserved in vertebrates35. Studies in mice revealed that Topaz1 plays a crucial role in spermatocyte, but not oocyte, progression through meiosis36. In men, TOPAZ1 is expressed in germ cells in both sexes31,37,38. Analysis of the testicular biopsy of this patient revealed a germ cell arrest in early spermiogenesis (Fig. 3).

a, b H&E stainings of (a) control and (b) Proband_060 with pathogenic mutations in TOPAZ1 gene. The epithelium of the seminiferous tubules in the TOPAZ1 proband show reduced numbers of germ cells and an absence of elongating spermatids based on the analysis of 150 seminiferous tubules in control and patient. c, d immunofluorescent labeling of DNA (magenta) and the acrosome (green) in control sections (c) and TOPAZ1 proband sections (d). (c) The arrowhead indicates the acrosome in an early round spermatid and the arrows the acrosome in elongating spermatids. Spreading of the acrosome and nuclear elongation are hallmarks of spermatid maturation. (d) No acrosomal spreading (see arrowheads) or nuclear elongation is observed in the TOPAZ1 proband. The asterisk indicates an example of progressive acrosome accumulation without spreading. Scale bar: 40 µm (a, b) and 5 µm (c, d).
Maternally inherited mutations can also result in dominant causes of male infertility if not affecting female fertility. We therefore studied all DNM genes for the presence of maternally inherited mutations in the entire cohort and compared this to the presence of paternally inherited mutations in the same genes. A total of 4 maternally inherited variants predicted to be pathogenic were identified in DNM genes (TENM2 (2×), CWC25, and EVC). All of these variants, however, were also observed multiple times in an exome dataset from a cohort of 5784 fertile men suggesting that these maternally inherited variants are not causative of male infertility (Supplementary Data 2).
Further analysis in additional cohorts of infertile males
In addition to all systematic analyses described above, we evaluated the function of all DNM genes to give each a final pathogenicity classification (Table 1, details in Methods). Of all 192 DNMs, 29 affected genes were linked to male reproduction and classified as possibly causative, with a further 50 as unclear. For replication purposes, only one pilot study including 13 trios was recently published in male infertility7. None of the DNM genes reported in this study showed DNMs in our cohort. To further study the DNM genes identified in our cohort, we looked for the presence of rare predicted pathogenic mutations in these genes in exome datasets of infertile men (n = 2,506), in collaboration with members of the International Male Infertility Genomics Consortium and the Geisinger-Regeneron DiscovEHR collaboration39. For comparison, we included an exome dataset from a cohort of 11,587 fertile men and women from Radboudumc.
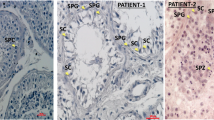
In the additional infertile cohorts, we identified 17 LoF mutations in our DNM LoF-intolerant genes (pLI ≥ 0.9), although we did not detect a statistical enrichment in the LoF mutations in these genes compared to fertile men (Two-tailed Fisher’s Exact test with Bonferroni correction adjusted p values >0.05, Supplementary Data 3, 4). Next, we looked for an enrichment of rare predicted pathogenic missense mutations in these cohorts (Table 2 and Supplementary Data 5, 6). A total of 11 genes showed an enrichment of pathogenic missense mutations in infertile men compared to fertile men (Two-tailed Fisher’s Exact test, p value < 0.05, Table 2). After applying the Bonferroni correction to counteract the effects of multiple testing, however, the only significant enrichment was observed in the RBM5 gene (adjusted p value = 0.03). In this gene, six infertile men were found to carry a rare pathogenic missense mutation, in addition to the proband with a de novo missense mutation (Supplementary Fig. 8, Supplementary Data 7). Importantly, no such predicted pathogenic mutations were identified in men in the fertile cohort. In line with these results, RBM5, already highlighted above as an essential regulator of male germ cell pre-mRNA splicing and male infertility26, is highly intolerant to missense mutations (missense Z-score 4.17).
In addition to the comparison between fertile and infertile men, we investigated whether there was any difference between the number of predicted pathogenic mutations carried in fertile men compared to fertile women. However, none of the DNM genes showed a significant difference between the sexes (Two-tailed Fisher’s Exact test with Bonferroni correction adjusted p values = 1, Supplementary Data 3, 5).
Phasing of de novo mutations to identify parent of origin
Given the predicted impact of these DNMs on spermatogenesis, we were interested in investigating the parental origin of DNMs in our trio cohort. We were able to phase 29% (n = 59) of all our DNMs using a combination of short-read WES and targeted long-read sequencing (Supplementary Table 1). In agreement with literature40,41,42,43, 72% of all DNMs occurred on the paternal allele. Interestingly, phasing of 8 likely causative DNMs showed that 6 of these were of paternal origin (75%). This suggests that DNMs with a deleterious effect on the future germline can escape negative selection in the paternal germline. This may be possible because the DNM occurred after the developmental window in which the gene is active, or the DNM may have affected a gene in the gamete’s genome that is critical for somatic cells supporting the (future) germline. Transmission of pathogenic DNMs may also be facilitated by the fact that from spermatogonia onwards, male germ cells form cysts and share mRNAs and proteins44. As such, the interconnectedness of male germ cells, which is essential for their survival45, could mask detrimental effects of DNMs occurring during spermatogenesis.
Discussion
In 2010, we published a pilot study pointing to a de novo paradigm for mental retardation46 (now more appropriately termed developmental disorders or intellectual disability). This work contributed to the widespread implementation of patient–parent WES studies in research and diagnostics for neurodevelopmental disorders47, accelerating disease gene identification and increasing the diagnostic yield for these disorders. The data presented here suggest that a similar benefit could be achieved from trio-based exome sequencing in male infertility. In order to achieve this there is an urgent need to expand on this work as larger studies are essential to identify recurrently DNM genes and further demonstrate the exact contribution of DNMs to male infertility. Modeling studies recently done for developmental disorders showed that more than 350k trios may be required to have approximately 80% power to detect all haploinsufficient genes causing this disorder48. Evidently, these numbers can only be reached by implementing trio-based exome sequencing as a routine diagnostic test and by sharing these diagnostic data with the international research community. This research community will also have the enormous task of functionally validating the impact of these DNMs on spermatogenesis. Altogether, this will not only help to increase the diagnostic yield for men with infertility but will also enhance our fundamental biological understanding of human reproduction and natural selection. In addition, it will indicate whether male infertility follows a dominant inheritance pattern, and this has impact for disease transmission. Couples that seek treatment for male infertility should be counseled on the risk of transmitting this condition to their offspring, something that is now limited to couples receiving fertility treatment due to Y-chromosome deletions. Male infertility is also increasingly seen as the most visible symptom of a more complex disease with associated comorbidities49. Studying the long-term health of men with DNMs in specific genes should help in identifying genotype–phenotype correlations that may impact more than the fertility of these men.
Methods
Cohort of infertile patients and fertile parent trios
We enrolled a total of 185 patients who presented with unexplained (idiopathic) azoospermia (N = 111) or severe to extreme oligozoospermia (with or without asthenozoospermia N = 74) at the Radboudumc outpatient clinic between July 2007 and October 2017 (N = 170) and at the Newcastle upon Tyne Hospitals NHS Foundation Trust (Newcastle, UK) between January 2018 to January 2020 (n = 15). The reference values and semen nomenclature were used according to the WHO guidelines50 (see Supplementary Note). Clinical evaluation did not lead to an etiologic diagnosis and all patients were negative for AZF deletions and chromosomal anomalies (see Supplementary Notes). The study protocol was approved by the respective Ethics Committees/Institutional Review Boards (Nijmegen: NL50495.091.14 version 5.0, Newcastle: REC ref. 18/NE/0089) and written informed consent from all patients and their parents was obtained prior to enrollment in the study. We used residual genomic DNA extracted from a blood sample taken at the time of evaluation and treatment at the fertility center. DNA from all proband’s parents was obtained from saliva by using the Oragene OG-500 kit (DNA Genotek, Ottawa, Canada).
Immunofluorescence staining of human testis biopsies
Tissue sections were cut from formalin fixed paraffin embedded (FFPE) testicular biopsies. As staining controls testicular biopsies obtained from fertile men after a previous vasectomy was used. FFPE sections were prepared for staining following standard protocols. To detect RBM5, antibody HPA018011 from Atlas Antibodies was used in a 1:500 dilution. For detection, a donkey anti-rabbit Alexa488 antibody was used (A-21206, 1:1000 dilution), which was applied in combination with lectin coupled to Alexa568 (L32458, 1:1500 dilution) to detect the acrosome (both Thermo Scientific). Slides were counterstained with DAPI. Images were obtained with a Zeiss Axio Imager ZI fluorescence microscope equipped with the Zen software package.
Cohort of verified fertile Dutch parents
We used an anonymized exome dataset derived from 5784 Dutch men and 5803 Dutch women who had conceived at least one child as a control cohort for the frequency of rare variants in fertile men and fertile women. These men and women received routine exome sequencing at the Radboud diagnostics center as the healthy parent of a child with a severe illness. Although these men fathered a child with intellectual disability, their fertility is expected to be similar to an unselected sample of the male population.
Exome sequencing
WES samples were prepared and enriched following the manufacturer’s protocols of either Illumina’s Nextera DNA Exome Capture kit or Twist Bioscience’s Twist Human Core Exome Kit. All sequencing was performed on the NovaSeq 6000 Sequencing System (Illumina) achieving comparable results covering more than 99% of all exonic regions using either kit (Supplementary Table 2) and an average depth of 72× (Illumina’s Nextera Kit) and 99x (Twist Bioscience’s Kit) (see Supplementary Fig. 9 and Supplementary Table 3). Sequenced reads were aligned to Human Reference Genome (GRCh37.p5/hg19) using BWA-Mem v0.7.1751, Picard52, and GATK v4.1.4.153. The sex, ancestry and relatedness of each samples was calculated using peddy54, samples found to have the incorrect sex or were unrelated to the correct samples were excluded from this study. Following best practice recommendations, single nucleotide variations and small indels were identified and quality-filtered using GATK’s HaplotypeCaller obtaining comparable results independently of the kit or the origin of the DNA (see Supplementary Table 3). Afterwards, all variants were further analysed using a custom GATK4-based algorithm to identify and separate high- and low-confidence de novo variants from inherited variants. Briefly, posterior genotype probabilities (GQ) were recalculated for each sample at each variant site using Bayes’ rule to take into account family and population priors53,55. Proband variants absent in parental samples with recalculated proband GQ > = 10 and allele count (AC) below 4 or allele frequency (AF) < 0.1% in all samples, whichever is more stringent, were classified as low-confidence DNMs. Variants with recalculated GQs ≥ 20 and the same AC/AF criterion were classified as high confidence DNMs. Afterwards, tagged variants with coverage <10, variant read percentage <15% and GATK quality scores <400 were removed to ensure only the most reliable variants were considered. Sanger sequencing was then used to validate DNMs calls. Ensembl’s Variant Effect Predictor (VEP)56 was used to fully annotate all de novo variants.
Variant filtration and interpretation
The primary stages in filtering of variants included removing all variants with an allele frequency of >0.1% in the gnomAD database to only include rare variants in our analysis. All variants then with <10 reads in the exome data and/or less than 15% of these reads containing the mutation were then removed. At this stage, any remaining variants lying outside the exonic regions were then removed. This provided the initial list of 192 rare de novo variants. All synonymous and non-protein-altering spice site variants were then removed, leaving a total of 145-protein-altering rare DNMs. Pathogenicity prediction was then based on SIFT20, MutationTaster21, and PolyPhen222 and all variants were classified according to the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) 2015 guidelines57. All protein-altering variants predicted to be pathogenic by at least 2 out of 3 prediction models, absent from the fertile male cohort, present in <5 males in the gnomAD database were considered for further functional analysis (n = 84). Maternally inherited mutations present in genes identified as having a protein-altering DNM were identified in all patients and submitted to the exact same method of filtration and interpretation as described above.
Functional analysis was split into six different categories, each category provided a score of either 1 or 0 depending on whether they met the threshold for that category. These categories included: RNA expression of the gene in the testis, RNA enrichment in the testis or presence in spermatogenesis, protein expression in the testis, whether an infertile mouse model already exists for the given gene, the protein function in relation to spermatogenesis and finally whether the given gene interacts with any known fertility genes. For expression levels retrieved for each gene of interest from the GTEx database (https://www.gtexportal.org/), an expression of medium (≥10 < 100 TPM) or high (>100 TPM) gave a score of 1 with low (>2 < 10 TPM) and no expression (<2 TPM) giving a score of 0. RNA enrichment was based on elevated expression (tissue enriched, group enriched, or tissue enhanced) in the Human Protein Atlas58 or being among the genes up- or downregulated during spermatogenesis as found in a recent single cell RNA sequencing study59. Protein expression was retrieved from the Human Protein Atlas58 and interaction with known infertility genes3 was calculated using STRING version 1124. The final classification of the genes was then split into Not causative, Unlikely causative, Unclear and Possibly causative. These classifications were given based on the variant scores out of 6 with: [0 points + not expressed/not detected/not present on several occasions = Unlikely causative], [0 points + “Unknown” on several occasions = Unclear], [1–2 points = Unclear] and [3–6 points = possibly causative].
CNV analysis
CNV calling was performed on our trio-based exome data with a custom GATK4-based pipeline. This workflow exploits the GATK4 sequence read depth normalization60 and a custom R based segmentation and visualization61. Parental samples from the trios under examination were used as controls for the normalization step. The CNVs detected were annotated using AnnotSV (https://lbgi.fr/AnnotSV/)62. CNVs present in more than 1% of the samples of the Database of Genomic Variants present in more than 10% of the patients were excluded from the analysis. The remaining rare deletions and duplications were individually inspected through the genomic profiles and detailed Log2Ratio plots generated by the workflow. Only CNVs involving more than 2 exons were further considered to minimize the inclusion of false positives, and we selected 2 CNVs present in the probands but absent in their parents for further validation.
Variant validation
Validation of low-quality DNMs was performed using standard Sanger sequencing approach on an Applied Biosystems SeqStudio Genetic Analyzer (ThermoFisher, MA, USA) to confirm the presence of the mutation in probands and its absence in the parents. Primers for each SNV were designed using PrimerZ63 (Supplementary Data 8) and PCR reactions were performed using AmpliTaq 360 DNA Polymerase (ThermoFisher, MA, USA) according to the manufacturer’s protocol.
Validation of CNVs was performed with the whole genome Illumina Infinium CytoSNP-850K v1.1 microarray platform for the larger deletion on chromosome 11 and a gene-specific TaqMan Copy-Number assay designed for NXT2 was exploited to validate the smaller CNV using the Applied Biosystems QuantStudio 7 Flex Real-Time PCR System (ThermoFisher, MA, USA).
Functional enrichment
To evaluate the intolerance of each gene for loss-of-function (LoF) mutations, we used the probability of LoF intolerance (pLI) score, based on data from the Genome Aggregation Database (gnomAD)8 containing genetic data from 141,456 individuals. We computed the likelihood of the observed median pLI score of each gene (LoF in controls) set compared to the expected median pLI based on the method described in Lelieveld et al.64. In short, we simulated the expected number of recurrently mutated genes by redistributing the observed number of mutations at random over a determined set of genes based on their specific LoF and functional mutation rates, however, in contrast to Lelieveld et al.64 and Samocha et al.23 before them instead of using the complete set of 18,226 pLI annotated genes to obtain expected median pLI scores, we used a set of 2766 coding DNMs in 1941 control individuals, downloaded from the de novo-db version 1.6.1 (http://de novo-db.gs.washington.edu/de novo-db/)12, to correct for the gene-specific mutation rate. The empirical P value was calculated by comparing the observed median pLI to the expected pLI following 100,000 random sampling simulations. Case and fertile controls were processed using the exact same filtration and annotation parameters as described above so that each variant detected was evaluated in a comparable manner. The same method was then repeated using the Loss-of-function observed/expected upper bound fraction or LOEUF score, which also is an indicator of LoF intolerance. To evaluate the impact of the de novo missense mutations to each gene, we used missense Z-scores calculated by gnomAD10,23 to predict the tolerance of each gene to variation in place of the pLI scores when applying the Lelieveld et al.64 methodology described above following 100,000 simulations. The presence of missense mutations in intolerant genes was compared between predicted pathogenic and benign using a two-tailed Mann–Whitney U test in our samples and in controls independently. To predict the affected protein function and the potential role in disease, we evaluated the interactions between the genes with a DNM using STRING version 1124.
Additional cohorts of infertile men
The strongest candidate genes with DNMs were further investigated in exome data from four additional cohorts of infertile men. For the Italian cohort of 48 patients with NOA, exome sequencing was carried out as a service by Macrogen Inc. (Republic of Korea) utilizing the Agilent SureSelect_V6 enrichment and a NovaSeq 6000. The German Male Reproductive Genomics (MERGE) study comprised exome data of 887 men with azoo-, crypto-, or severe oligozoospermia. Known causes for male infertility like chromosomal aberrations and microdeletions of the AZF region were excluded in advance. WES was performed as previously described65. The 88 patients diagnosed with male infertility participating in the Geisinger-Regeneron DiscovEHR collaboration were selected from deidentified EHR information using the ICD-10CM code N46 which refers to “Male Infertility” including oligospermia, azoospermia, other male infertility and male infertility unspecified. All patients were sequenced at the Regeneron Genetics Center (RGC) as previously described39. In brief, 1ug of genomic DNA per sample was used for targeted exome capture using the NimbleGen VCRome 2.1 or the IDT XGen reagents. Captured libraries were sequenced on the Illumina HiSeq 2500 platform with v4 chemistry using paired-end 75 bp reads. Exome sequencing was performed such that >85% of the bases were covered at 20× or greater. Raw sequence reads were mapped and aligned to the GRCh38/hg38 human genome reference assembly using BWA-mem51, single nucleotide and indel variants were called using GATK’s HaplotypeCaller53. The Genetics of Male Infertility INitiative (GEMINI) is a multicenter study funded by the United States NIH. The GEMINI project performed whole-exome sequencing on 1,011 unrelated men diagnosed with spermatogenic failure, the vast majority with unexplained NOA. Sequencing of genomic DNA was performed at the McDonnell Genome Institute of Washington University in St. Louis, MO, USA, using an in-house exome targeting reagent capturing 39.1 Mb of exome and 2 × 150 bp paired-end sequencing on Illumina HiSeq 4000. Following sample QC, a final cohort of 924 men were analysed as part of the current study.
Genetic variants identified within the 152 candidate genes were extracted from each exome dataset. Consistent with our filtering method described above variants with <10 reads and/or <15% reads containing the mutation were discarded. To minimize discrepancies between genomic positions and annotation, genomic coordinates were recalculated to the GCRh37/hg19 where necessary and fully reannotated with VEP56. Following annotation variants from each of the additional case and control cohorts were filtered and processed in an identical manner as previously described. Shortly, variants with allele frequency >1% in gnomAD were discarded to focus only on rare variants: Pathogenicity predictions based on SIFT20, MutationTaster21 and PolyPhen22 were then used to exclude benign variants, all remaining variants were classified according to ACMG guidelines56. Like before, all protein-altering variants were considered pathogenic if predicted to be so by at least 2 out of 3 prediction models, absent from the fertile cohorts and present in <5 males in the gnomAD database. A similar analysis was done for all variants obtained in the control cohorts. However, just to be clear, for the male control cohort we did not exclude variants as pathogenic if they were present in this cohort itself.
Burden testing
Having identified several likely pathogenic rare loss-of-function and missense mutations in these 152 genes we performed a gene-based burden test to compare the combined data in all cohorts of infertile men with the control cohort of fertile fathers. The proportion of individuals with pathogenic variants in each of the 152 genes was statistically evaluated using two-tailed Fisher’s Exact tests, individual p values were corrected using the Bonferroni method corrections to adjust for performing 152 consecutive statistical tests and reduced the risk of Type I errors. Similarly, a gene-based burden test was performed to compare fertile fathers with fertile mothers from the control cohort of verified fertile parents to investigate whether any of the sexes predominantly carried a greater number of rare pathogenic mutations.
Phasing analysis to determine parent-of-origin
The origin of DNMs identified in the exomes of patients was first investigated in the short-read exome data by performing phasing analysis on those variants that contained a parental informative SNP (iSNP) within 150 bp from the DNM. As a next step, all DNMs were target-enriched with long-range PCR and sequenced using the Oxford Nanopore’s MinION sequencer (Oxford Nanopore technologies, Oxford Science Park, UK). Target regions were designed to encapsulate both the DNM and a parentally informative SNP, from which parent-of-origin, and allele frequencies (percentage read counts associated to a given allele) could be ascertained and DNM pre-/post-zygosity could be determined.
Primers were designed using Primer366 (version 2.3.6) and GRCh37.p5 based in-house GUI-wrapped pipeline. All expected fragment sizes were limited to a maximum of 12 kb for quality control and enrichment success rate. For those DNMs with no exome supported iSNPs within a 10 kb distance, primers were designed to cover approximately 2.5 kb on either side of the DNM with the expectation of finding additional iSNPs in the intronic regions. Long-range PCR target enrichment was carried out using our optimized running conditions of 3 separate supermixes/enzymes (see Supplementary Table 4). Sample fragment sizes were confirmed using gel electrophoresis, and quantities were measured with the Qubit dsDNA HS kit (ThermoFisher Scientific, Waltham, MA, USA), with the best quality supermix enrichment for each given sample/target selected for sequencing, where quality was assessed by cleanest banding in gel electrophoresis and greatest concentration.
The long-range PCR target enrichments of >20 ng were prepared for sequencing with the ONT ligation sequencing kit (SQK-LSK109) following the manufacturer’s protocol, with adjustments for sample type and yield. Individual sample libraries were concentrated where necessary at given bead clean-up steps and pooled based on fragment size. Fragment size-based pools were combined prior to flowcell loading. Prepared samples were sequenced on the MinION using the FLO-MIN106 version 9.4 flowcell platform. Flowcells were run until complete pore exhaustion, with minimal refuel of flowcells performed whenever active pore percentages dropped below 70%, achieving an average of 30 billion basecall yields per flowcell and coverage depth per sample of >5000×.
The sequence signal data in multi-fast5 format were basecalled using Guppy67 (version 3.4.4, https://nanoporetech.com/), resulting fastq outputs were adapter trimmed and low-quality reads discarded using cutadapt (version 2.5)68. Cleaned fastq files were mapped against Human Reference Genome (GRCh37.p5/hg19) using BWA-Mem51 (version 0.7.17), and sample targets were extracted from the resulting BAM file using SAMtools (version 0.1.19)69. Aligned ONT reads were phased using an in-house tool, with frequencies and pre/post-zygosity calls affirmed via IGV and principal component analysis using the available exome sequence data for probands and parents to support the ONT data.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Sequencing data has been deposited in the European Genome-phenome Archive(EGA) under the accession code EGAS00001005417 and will be made available upon reasonable request for academic use and within the limitations of the provided informed consent by the corresponding author upon acceptance. Every request will be reviewed by the Newcastle University Male Infertility Genomics Data Access Committee; the researcher will need to sign a data access agreement after approval.
Code availability
The code for the integrated pipeline used to process sequencing data to detect and call rare germline copy-number variants (CNVs) is available at https://github.com/AnetaMikulasova/CNVRobot.
References
Krausz, C. & Riera-Escamilla, A. Genetics of male infertility. Nat. Rev. Urol. 15, 369–384 (2018).
Tüttelmann, F., Ruckert, C. & Röpke, A. Disorders of spermatogenesis. Med. Genet. 30, 12–20 (2018).
Oud, M. S. et al. A systematic review and standardized clinical validity assessment of male infertility genes. Hum. Reprod. 34, 932–941 (2019).
Kasak, L. & Laan, M. Monogenic causes of non-obstructive azoospermia: challenges, established knowledge, limitations and perspectives. Hum. Genet. 140, 135–154 (2021).
Veltman, J. A. & Brunner, H. G. de novo mutations in human genetic disease. Nat. Rev. Genet. 13, 565–575 (2012).
Sun, C. et al. An azoospermic man with a de novo point mutation in the Y-chromosomal gene USP9Y. Nat. Genet. 23, 429–432 (1999).
Hodžić, A. et al. de novo mutations in idiopathic male infertility—a pilot study. Andrology 9, 212–220 (2021).
De Jonge, C. & Barratt, C. L. R. The present crisis in male reproductive health: an urgent need for a political, social, and research roadmap. Andrology 7, 762–768 (2019).
Schilit, S. L. P. et al. SYCP2 translocation-mediated dysregulation and frameshift variants cause human male infertility. Am. J. Hum. Genet. 106, 41–57 (2020).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
denovo-db, Seattle, WA. http://denovo-db.gs.washington.edu (2020).
Gu, Y. et al. Three intellectual disability-associated de novo mutations in MECP2 identified by trio-WES analysis. BMC Med. Genet. 21, 99 (2020).
Fritzen, D. et al. de novo FBXO11 mutations are associated with intellectual disability and behavioural anomalies. Hum. Genet. 137, 401–411 (2018).
De Tomasi, L. et al. Mutations in GREB1L cause bilateral kidney agenesis in humans and mice. Am. J. Hum. Genet. 101, 803–814 (2017).
Brophy, P. D. et al. A gene implicated in activation of retinoic acid receptor targets is a novel renal agenesis gene in humans. Genetics 207, 215–228 (2017).
Alhathal, N. et al. A genomics approach to male infertility. Genet. Med. 22, 1967–1975 (2020).
Seabra, C. M. et al. A novel Alu-mediated microdeletion at 11p13 removes WT1 in a patient with cryptorchidism and azoospermia. Reprod. Biomed. Online 29, 388–391 (2014).
Grozdanov, P. N., Li, J., Yu, P., Yan, W. & MacDonald, C. C. Cstf2t regulates expression of histones and histone-like proteins in male germ cells. Andrology 6, 605–615 (2018).
Vaser, R., Adusumalli, S., Leng, S. N., Sikic, M. & Ng, P. C. SIFT missense predictions for genomes. Nat. Protoc. 11, 1–9 (2016).
Schwarz, J. M., Rödelsperger, C., Schuelke, M. & Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576 (2010).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Samocha, K. E. et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 46, 944–950 (2014).
Szklarczyk, D. et al. The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362–D368 (2017).
Song, H., Wang, L., Chen, D. & Li, F. The function of Pre-mRNA alternative splicing in mammal spermatogenesis. Int. J. Biol. Sci. 16, 38–48 (2020).
O’Bryan, M. K. et al. RBM5 is a male germ cell splicing factor and is required for spermatid differentiation and male fertility. PLoS Genet. 9, e1003628 (2013).
Elsea, S. H. & Lucas, R. E. The mousetrap: what we can learn when the mouse model does not mimic the human disease. ILAR J. 43, 66–79 (2002).
Jamsai, D. et al. In vivo evidence that RBM5 is a tumour suppressor in the lung. Sci. Rep. 7, 16323 (2017).
Liang, H. et al. Differential Expression of RBM5, EGFR and KRAS mRNA and protein in non-small cell lung cancer tissues. J. Exp. Clin. Cancer Res. 31, 36 (2012).
Li, J. et al. HnRNPL as a key factor in spermatogenesis: Lesson from functional proteomic studies of azoospermia patients with sertoli cell only syndrome. J. Proteomics 75, 2879–2891 (2012).
Wang, M. et al. Single-Cell RNA sequencing analysis reveals sequential cell fate transition during human spermatogenesis. Cell Stem Cell 23, 599–614.e4 (2018).
Ajuh, P. Functional analysis of the human CDC5L complex and identification of its components by mass spectrometry. EMBO J. 19, 6569–6581 (2000).
Brea-Fernández, A. J. et al. Expanding the clinical and molecular spectrum of the CWC27-related spliceosomopathy. J. Hum. Genet. 64, 1133–1136 (2019).
Millevoi, S. et al. An interaction between U2AF 65 and CF Im links the splicing and 3′ end processing machineries. EMBO J. 25, 4854–4864 (2006).
Baillet, A. et al. TOPAZ1, a novel germ cell-specific expressed gene conserved during evolution across vertebrates. PLoS ONE 6, e26950 (2011).
Luangpraseuth-Prosper, A. et al. TOPAZ1, a germ cell specific factor, is essential for male meiotic progression. Dev. Biol. 406, 158–171 (2015).
Guo, F. et al. The transcriptome and DNA methylome landscapes of human primordial germ cells. Cell 161, 1437–1452 (2015).
Li, L. et al. Single-cell RNA-seq analysis maps development of human germline cells and gonadal niche interactions. Cell Stem Cell 20, 858–873.e4 (2017).
Dewey, F. E. et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science. 354, aaf6814 (2016).
Francioli, L. C. et al. Genome-wide patterns and properties of de novo mutations in humans. Nat. Genet. 47, 822–826 (2015).
Rahbari, R. et al. Timing, rates and spectra of human germline mutation. Nat. Genet. 48, 126–133 (2016).
Goldmann, J. M. et al. Parent-of-origin-specific signatures of de novo mutations. Nat. Genet. 48, 935–939 (2016).
Jónsson, H. et al. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature 549, 519–522 (2017).
Braun, R. E., Behringer, R. R., Peschon, J. J., Brinster, R. L. & Palmiter, R. D. Genetically haploid spermatids are phenotypically diploid. Nature 337, 373–376 (1989).
Greenbaum, M. P., Iwamori, T., Buchold, G. M. & Matzuk, M. M. Germ cell intercellular bridges. Cold Spring Harb. Perspect. Biol. 3, a005850–a005850 (2011).
Vissers, L. E. L. M. et al. A de novo paradigm for mental retardation. Nat. Genet. 42, 1109–1112 (2010).
Vissers, L. E. L. M., Gilissen, C. & Veltman, J. A. Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 17, 9–18 (2016).
Kaplanis, J. et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature. 586, 757–762 (2020).
Kasman, A. M., Del Giudice, F. & Eisenberg, M. L. New insights to guide patient care: the bidirectional relationship between male infertility and male health. Fertil. Steril 113, 469–477 (2020).
World Health Organization. WHO laboratory manual for the examination and processing of human semen. vol. 5th ed. (World Health Organization, 2010).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010).
Broad Institute. Picard toolkit. https://broadinstitute.github.io/picard/ (2019).
Auwera, G. A. et al. From FastQ data to high‐confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 43, 11.10.1–11.10.33 (2013).
Pedersen, B. S. & Quinlan, A. R. Who’s Who? Detecting and Resolving Sample Anomalies in Human DNA Sequencing Studies with Peddy. Am J Hum. Genet. 100, 406–413 (2017).
Poplin, R. et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 201178. https://doi.org/10.1101/201178 (2018).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol 17, 122 (2016).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423 (2015).
Uhlén, M. et al. Tissue-based map of the human proteome. Science 347, 1260419 (2015)
Guo, J. et al. The adult human testis transcriptional cell atlas. Cell Res. 28, 1141–1157 (2018).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing,Vienna, Austria. https://www.R-project.org/ (2017).
Geoffroy, V. et al. AnnotSV: an integrated tool for structural variations annotation. Bioinformatics 34, 3572–3574 (2018).
Tsai, M.-F. et al. PrimerZ: streamlined primer design for promoters, exons and human SNPs. Nucleic Acids Res. 35, W63–W65 (2007).
Lelieveld, S. H. et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci. 19, 1194–1196 (2016).
Wyrwoll, M. J. et al. Bi-allelic mutations in M1AP are a frequent cause of meiotic arrest and severely impaired spermatogenesis leading to male infertility. Am. J. Hum. Genet. 107, 342–351 (2020).
Untergasser, A. et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 40, e115 (2012).
Wick, R. R., Judd, L. M. & Holt, K. E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 20, 129 (2019).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J. 17, 10–12 (2011).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Acknowledgements
We are grateful for the participation of all patients and their parents in this study. We thank Laurens van de Wiel (Radboudumc), Sebastian Judd-Mole (Monash University), Arron Scott and Bryan Hepworth (Newcastle University) for technical support, and Margot J Wyrwoll (University of Münster) for help with handling MERGE samples and data. This project was funded by The Netherlands Organization for Scientific Research (918-15-667) to J.A.V. as well as an Investigator Award in Science from the Wellcome Trust (209451) to J.A.V. a grant from the Catherine van Tussenbroek Foundation to M.S.O. a grant from MERCK to R.S. a UUKi Rutherford Fund Fellowship awarded to B.J.H. and the German Research Foundation Clinical Research Unit “Male Germ Cells” (DFG, CRU326) to C.F. and F.T. This project was also supported in part by funding from the Australian National Health and Medical Research Council (APP1120356) to M.K.O.B., by grants from the National Institutes of Health of the United States of America (R01HD078641 to D.F.C. and K.I.A., P50HD096723 to D.F.C.) and from the Biotechnology and Biological Sciences Research Council (BB/S008039/1) to D.J.E.
Author information
Authors and Affiliations
Consortia
Contributions
This study was designed by M.S.O., L.E.L.M.V., L.R. and J.A.V. R.M.S., J.G., H.T. and G.W.v.d.H. provided all clinical data and performed the TESE histology and cytology analysis under supervision of L.R., D.D.M.B., E.S., K.F., K.D.H. and K.M. J.C. performed the exome sequencing with support from B.A. and bioinformatics support was provided by M.J.X., G.A., C.G. and S.C. Sanger sequencing was performed by P.F.d.V., H.I., H.E.S., L.E.B. and B.K.S.A. M.S.O. and H.E.S. performed the SNV analyses with support from M.J.X., F.K.M. performed CNV analysis with support from A.M. and M.S.K. and G.S.H. and L.E.B. performed the phasing. D.J.E., H.S., B.J.H. and M.K.O.B. provided support on the functional interpretation of mutations. D.F.C., L.N., C.F., S.K., F.T., K.I.A., A.R.E., C.K. and C.G.-J. were involved in the replication study. The first draft of the paper was prepared by M.S.O., H.E.S., R.M.S., M.J.X., G.W.v.d.H. and J.A.V.. All authors contributed to the final paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Oud, M.S., Smits, R.M., Smith, H.E. et al. A de novo paradigm for male infertility. Nat Commun 13, 154 (2022). https://doi.org/10.1038/s41467-021-27132-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-27132-8
This article is cited by
-
Low androgen signaling rescues genome integrity with innate immune response by reducing fertility in humans
Cell Death & Disease (2024)
-
Frequency, morbidity and equity — the case for increased research on male fertility
Nature Reviews Urology (2024)
-
DDX3Y is likely the key spermatogenic factor in the AZFa region that contributes to human non-obstructive azoospermia
Communications Biology (2023)
-
Large-scale analysis of de novo mutations identifies risk genes for female infertility characterized by oocyte and early embryo defects
Genome Biology (2023)
-
Diverse monogenic subforms of human spermatogenic failure
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
